Underestimated kmer-based genome size estimate: supperrepetitive sequences
The marbled crayfish Procambarus virginalis is one of the very young asexual species, probably created by aquarists in Germany about 60 years ago. Since then the species became invasive and expanded its range all the way up to Madagascar. This asexual species is a triploid, suposedly derived from its sexual diploid sister species Procambarus fallax. This peculiar genome has a known karyotype and well measured genome size using flow cytometry. We therefore have a good expectation of the haploid genome size (~3.5Gbp).
The genome was recently sequenced in Gutekunst et al. 2018 and assembled in very similar span to the genome size expectation. The data were collected and reanalyzed for comparison with other asexual species in our recent preprint.
I worked on this observation with Timothy Rhyker Ranallo-Benavidez with a great help from Julian Gutekunst.
Observation
We used default genome profiling to characterize the heterozygosity of the species. However, we found that the model underestimates the genome size by more than one gigabase
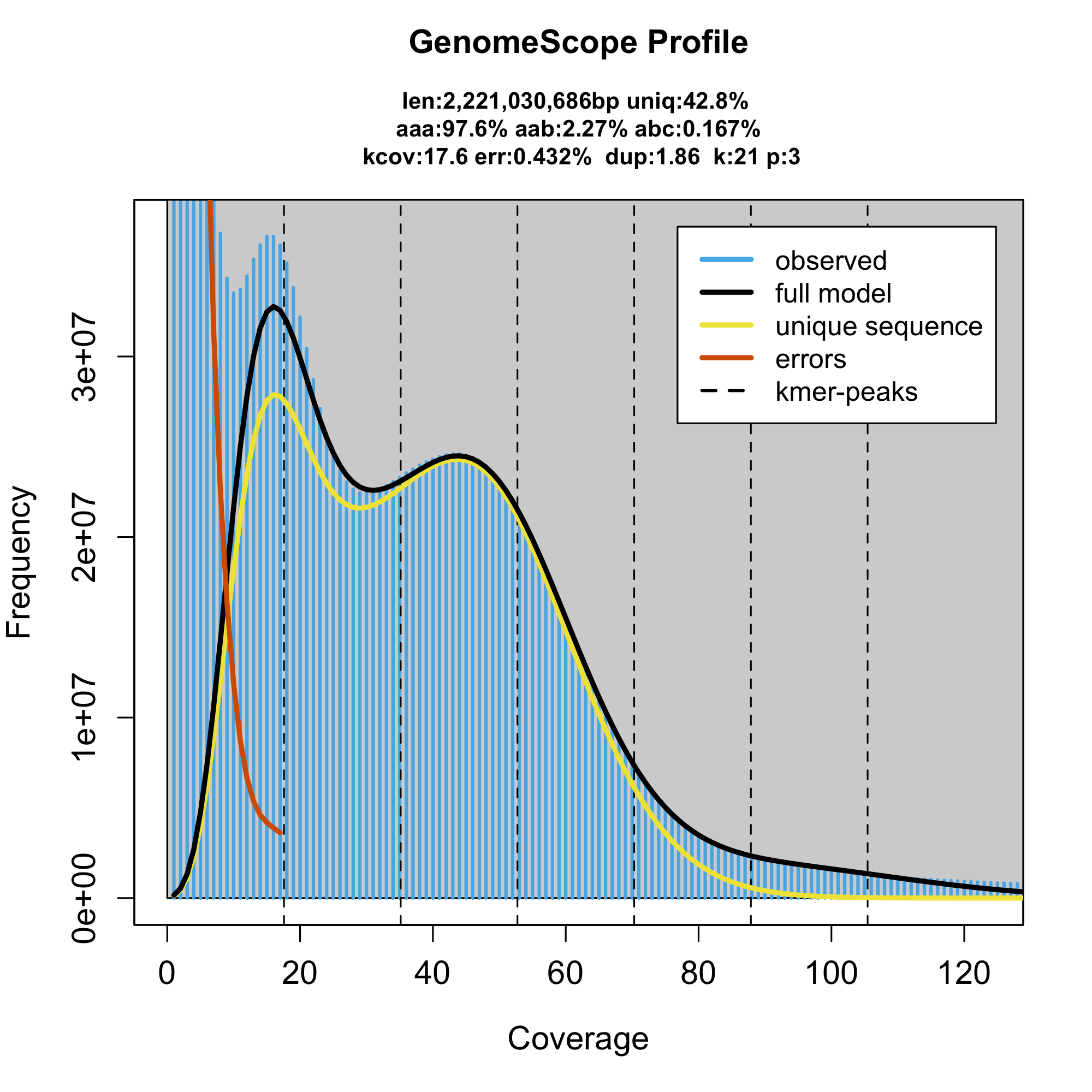
The genome assembly had the correct genome size, therefore we suspected that problem must be introduced somewhere on our side. As the genome size estimated only as a sum of all the genomic kmers divided by ploidy and haploid genome coverage, there are only two possible problems. Either the haploid coverage or the sum are wrong. While the coverage was in agreement with report in the genome assembly, the sum of kmer coverages was smaller than expected. Then we realized that the kmer counter we used (jellyfish) stops counting when it reaches coverage 10000, as kmers with this and higher are unlikely real genomic kmer, but rather adapters or contaminants. However, that applies only for small compact genomes, the crayfish repetitiveness might be, and it is, much higher. When we relaxed this constrained as requested the counter to generate histogram of all the coverages explicitly, we fit the following model
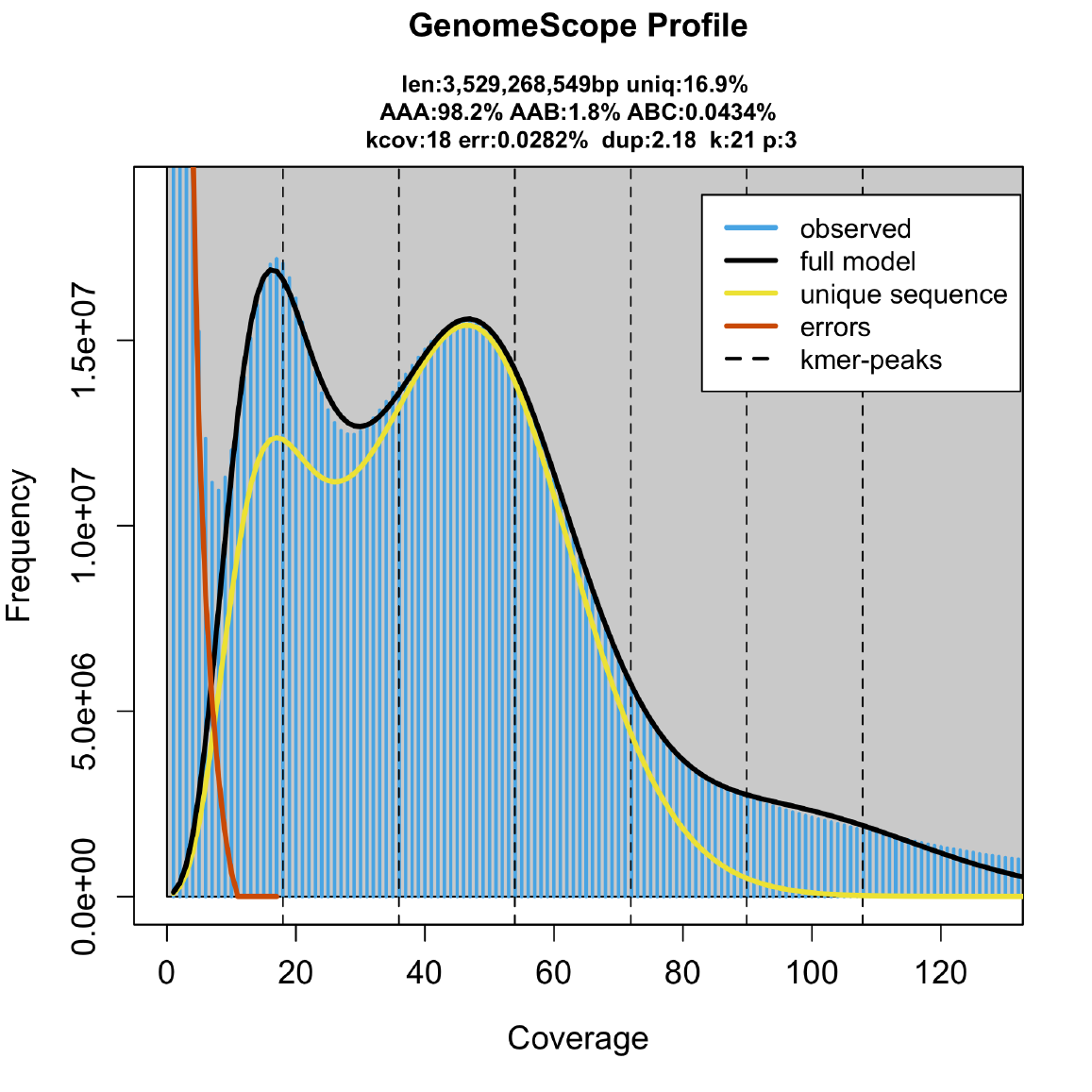
The genome size fits perfectly.
Explanation
Taking in consideration both models it suggest that more than a gigabase of the crayfish genome is actually covered by these supperrepetitive sequences. Without knowing what they are, we estimated that the most abundant 21 bases in the genome is present in approximately 7,280,000 genomic copies.
Within the same paper we have also estimated transposable element abundance directly genome genomic reads and their ages and indeed, there seems to be a recent expansion of long terminal repeats and several LINE families
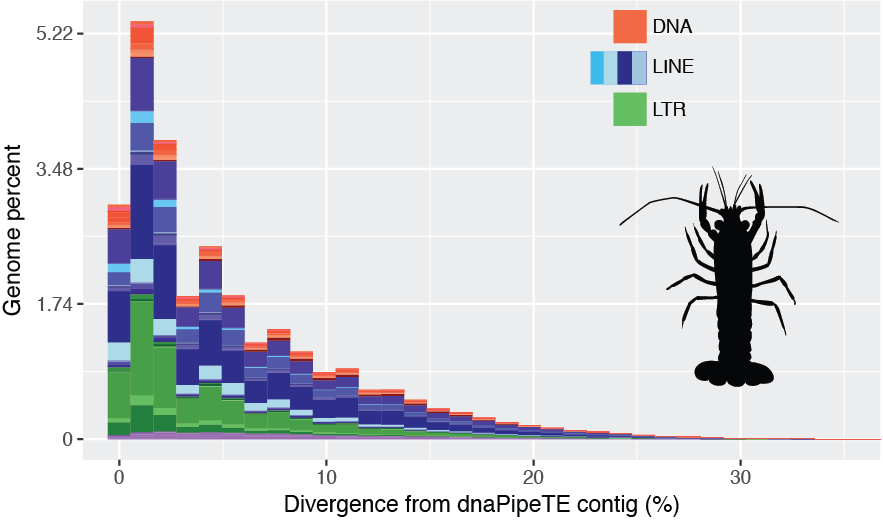
Although we don’t know if the most abundant sequence is an actual transposable element, it’s almost certain that the recent expansion significantly contribute to the supperrepetitive nature of the crayfish genome.
Just for the record, although the genome assembly has the correct span, nearly half of the bases are Ns, therefore only 4% of the genome were annotated as repeats.
Methods
Used data:
- the crayfish sequencing (SRA ids:
SRR5115143,SRR5115144,SRR5115145,SRR5115146,SRR5115147,SRR5115148)
Used software:
- KMC to generate kmer histogram
- GenomeScope for genome profiling
- dnaPipeTE for the TE profile
Genome profiling
Taken from our recent preprint, but it is basically the very default genome profiling analysis.
# dl SRR8061556.sra file; convert it to fastq files; erase the .sra file
ls *.fastq > FILES
kmc -k21 -t16 -m64 -ci1 -cs10000 @FILES kmer_counts tmp
kmc_tools transform kmer_counts histogram kmer_k21.hist -cx10000
genomescope.R -i kmer_k21.hist -o output_dir -n crayfish
then the corrected analysis allowed counting of the coverage up to 500,000,000x. Technical note: kmc_tools prints counts of every coverage till the specified including the 0 counts. The awk line just extracts all the coverages with non-zoro count to make the histogram smaller.
kmc -k21 -t16 -m64 -ci1 -cs500000000 @FILES kmer_counts tmp
kmc_tools transform kmer_counts histogram kmer_k21_full_with_zeros.hist -cx500000000
awk '{ if( $2 != 0 ){ print $0 } }' kmer_k21_full_with_zeros.hist > kmer_k21_full.hist && rm kmer_k21_full_with_zeros.hist
genomescope.R -i kmer_k21_full.hist -o output_dir -n crayfish_superrepetitive